27. Energies of stable conformers in heavy alkanes and triglycerides
27.1. Purpose
This application note demonstrates the use of molecular simulations to improve the understanding of structure and energetics of conformers [1] of heavy alkanes and triglycerides.
Understanding organic molecular chain conformations and computing their relative energies is of critical importance for materials science and engineering due to the significant impact on physical and chemical properties.
In life-science, conformations determine even the biological activity of drug molecules.
27.2. Method
Our method of choice for this application is the semi-empirical quantum chemistry (QM) code MOPAC [2] as implemented in the MedeA® Environment [3]. Semi-empirical QM methods, such as MOPAC, achieve considerable speed-up and gain in accuracy over traditional Hartree-Fock quantum chemistry methods by using empirical (=derived from an experiment) corrections. MedeA MOPAC uses the so-called PM7 approximation developed specifically for use with large organic molecules [4].
27.3. Alkanes and Triglycerides
Both alkanes and triglycerides contain flexible chains that can adopt many different conformations depending on the local environment and temperature [5], [6]. Linear alkanes are the principal components of crude oils, condensate gases, fuels, and waxes [7]. Triglycerides are the main compounds of vegetable oils and fats, and they are used by living organisms to store solar energy in a very compact form [8]. A better understanding of their properties has significant implications for power and food supply at the global level.
27.3.1. Alkane structure
Under standard conditions (25 oC and 1 atm), pure n-alkanes with up to 4 carbon atoms are gaseous, with 5 to 16 carbon atoms are liquid and with more than 16 carbon atoms are solid in the form of paraffin crystals.
N-alkanes consist of two methyl CH3 end-groups connected by a chain of methylene CH2 groups. Typical features of alkyl chains are an average C-C bond length of 1.54 \({\mathring{\mathrm{A}}}\) and C-C-C angles of 114 o. Alkyl chains can adopt many different conformations, depending on the dihedral torsion angles of each four successive carbons in the chain. Each dihedral angle may take one of three stable positions: trans (T), gauche counterclockwise (G), or gauche clockwise (G’).
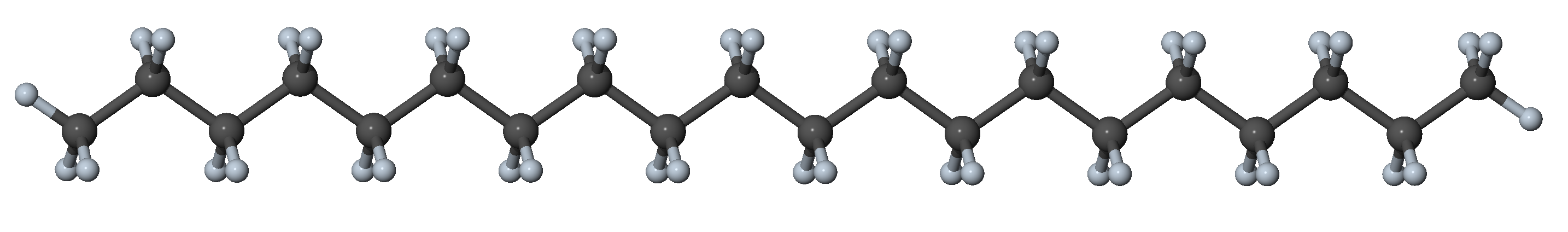
Figure 27.3.1.1 N-alkane in the all-trans conformation
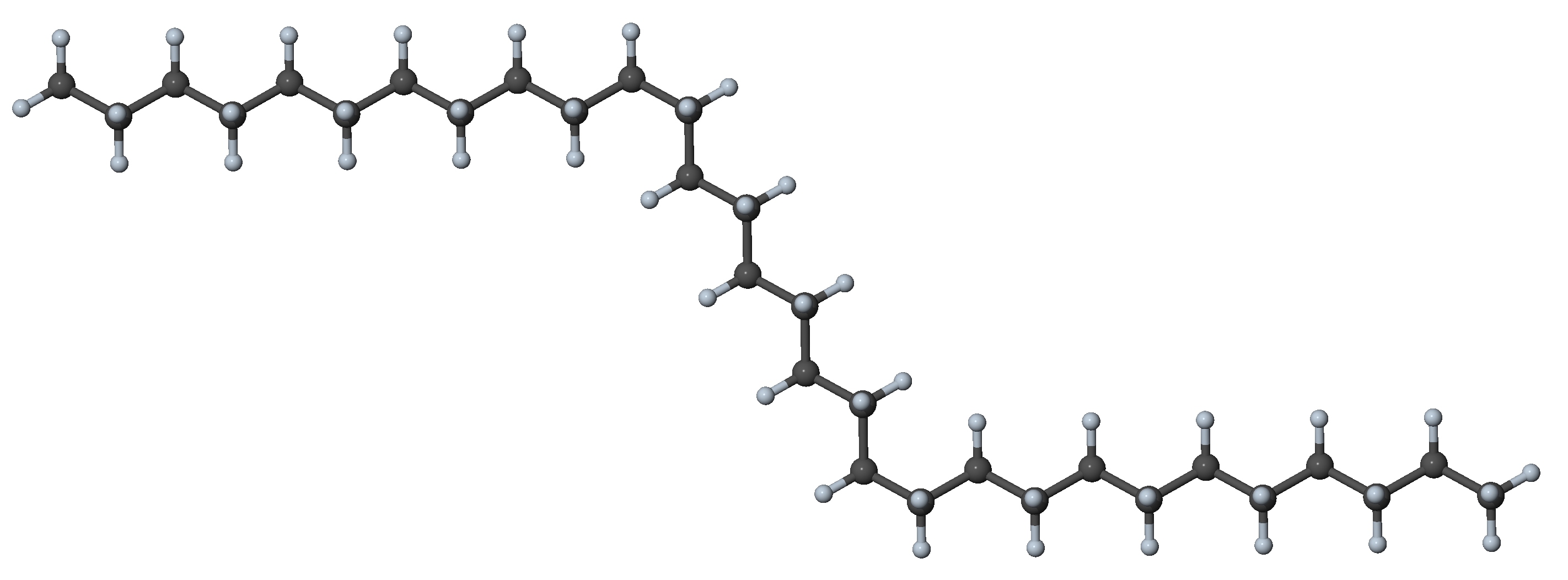
Figure 27.3.1.2 N-alkane with two gauche dihedrals and all other dihedral angles in trans conformation
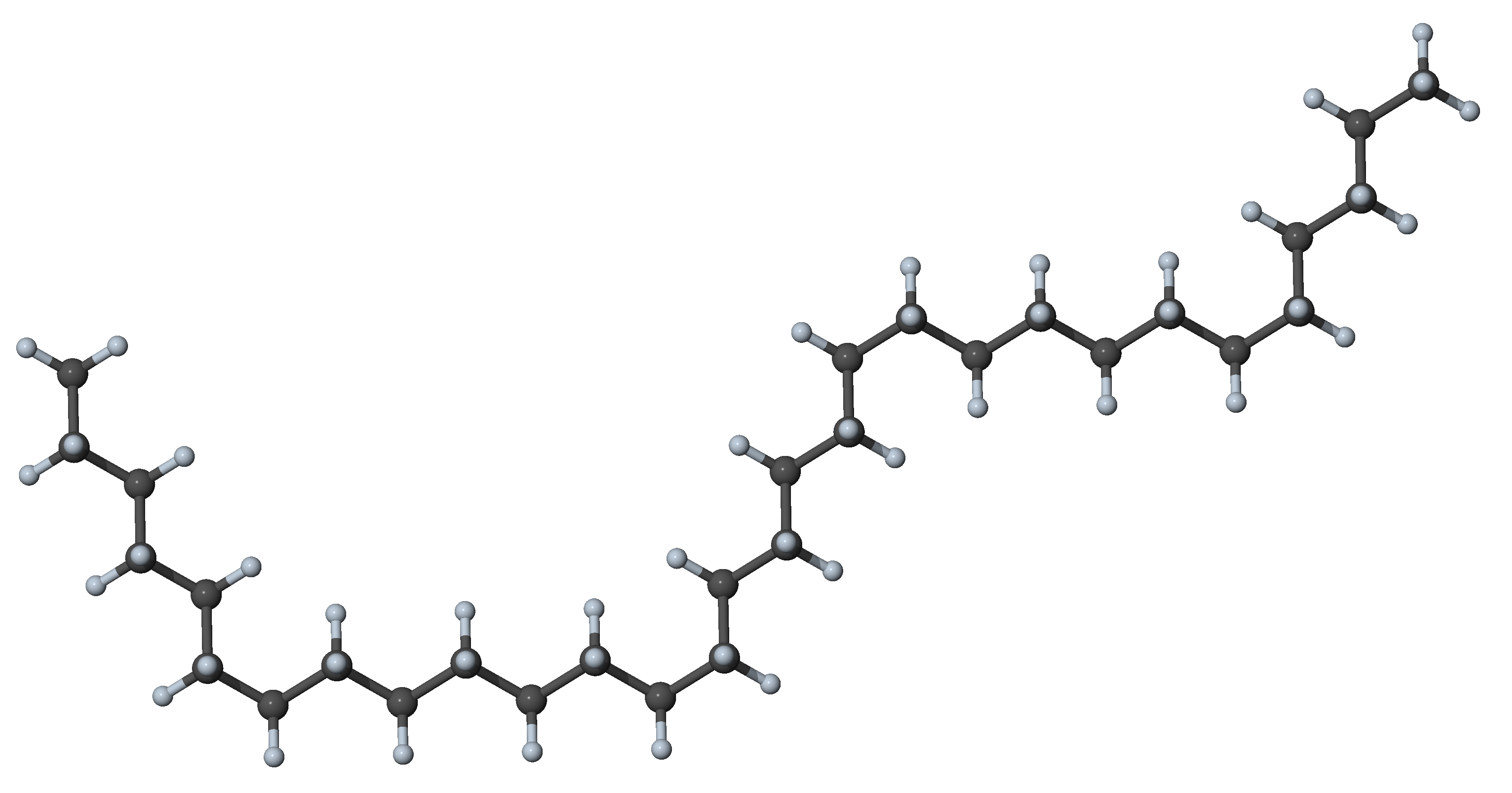
Figure 27.3.1.3 N-alkane with four gauche dihedrals and other dihedral angles in trans position, a conformer with little interaction between segments

Figure 27.3.1.4 N-alkane with four gauche dihedrals and all other dihedrals in trans position: hairpin conformer with significant interaction between segments

Figure 27.3.1.5 N-alkane with eight gauche dihedrals and all other dihedrals in trans position: double-hairpin conformer with significant interaction between segments
In n-alkanes with up to ~18 carbon atoms, the all-trans configuration is the most stable one [5]. With every gauche dihedral angle, the potential energy increases by ~2-3 kJ mol-1 [5]. While gauche conformers are energetically less favorable, they are present under ambient conditions, because small amounts of thermal energy are sufficient to overcome the energy barriers between conformers.
In n-alkanes with more than 20 carbon atoms, the most stable form is no longer the all-trans conformer, and folded conformers prevail. In these folded conformers, the attraction between adjacent chains compensates the energy cost for the gauche dihedrals required to bend the chain. Because the alkanes are non-polar, the attraction between the chains is mostly due to dispersion forces, which decay fast (1/r 6) with distance [9]. Several folded stable conformers of heavy alkanes have names that allude to the geometry of the folding such as hairpin, bundle, paperclip, among others [5].
27.3.2. Triglycerides
Most triglycerides of biological origin are esters of glycerol in which three alkyl chains link with the glycerol skeleton, each through an ether group [8]. The chains bear the name of the corresponding fatty acid, such as palmitic or stearic. Plants synthesize alkyl chains preferentially with an even number of carbons, the most common being 16 or 18 carbons.

Figure 27.3.2.1 Example of folded stable triglyceride conformer with 57 carbon atoms (3 saturated chains of 18 carbons).
27.4. Molecular modeling
The MedeA Environment is used for building representative structure models with up to 60 carbon atoms for the two species, and for performing quantum chemistry simulations to determine their energies.
In MedeA, we build all molecules using a SMILES string [10], and using the conformer search tool, we automatically generate a subset of all possible conformations, along with a forcefield-based estimation of each conformer energy. We store the resulting conformers in a structure list as input for use with any of MedeA’s compute engines. For low and medium molecular-weight n-alkanes, this approach is very efficient in generating stable conformers with different dihedral angles. However, because the number of conformers increases as 3n-3, where n is the number of carbons in the alkyl chain, a systematic search is feasible only for up to n~12. Beyond n~20, the huge number of possibilities prohibits a systematic search.
For exploring energetically favorable conformers of larger molecules, forcefield-based methods (Monte Carlo or Molecular dynamics) can be considered [5]. However, additional quantum mechanics calculations are required to predict the energy contribution of the vibrational degrees of freedom.
Here, MedeA MOPAC is used to find the structure and properties of stable conformers as it explicitly accounts for dispersion energy [4].
27.5. Results
Figure 27.5.1 displays the trend of energies for various gauche conformers of n-alkanes, including unfolded and folded conformers. For shorter chain lengths, a few points computed with high accuracy (ab-initio) quantum-chemistry methods, including dispersion interactions [6], are shown for comparison. We note that MOPAC produces very similar trends, correctly indicating the change toward negative folding energies in the C13-C18 range: The longer the parallel segments of the hairpin, the larger the energy difference with the all-trans conformer. For alkanes with chain length C16 to C22, where data from higher-accuracy methods are available, we note a systematic deviation of MOPAC results by less than 10 kJ mol-1, in agreement with the expected accuracy of the MOPAC PM7 parametrization [4].
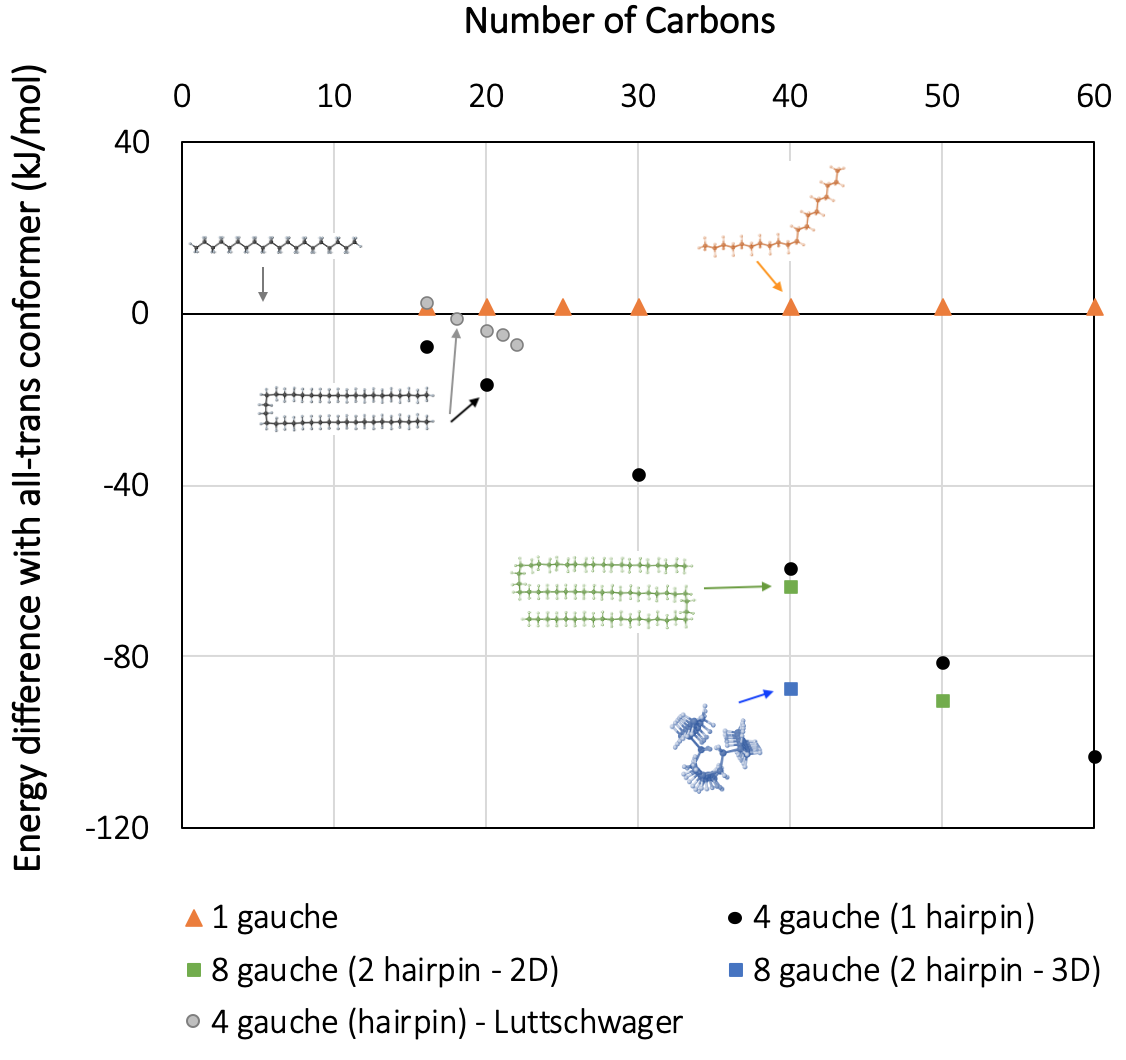
Figure 27.5.1 Standard enthalpy difference between several linear alkane conformers and the corresponding all-trans conformer with 16 to 60 carbon atoms, using MedeA MOPAC. For shorter chain lengths, high-accuracy quantum-chemistry data from the literature (MP2-CC and B3LYP-D3-TZVP) are reported for comparison [6].
With MedeA MOPAC, geometry optimizations using PM7 are performed up to chain lengths of n=60 with a modest computational expense, while ab-initio methods become very slow and unmanageable in this range. Our results indicate that for long-chain alkanes, the folded conformers containing two GGTGG sequences (also known as a “bundle” conformation) are energetically more favorable. A plausible explanation for this behavior is that the third straight chain segment interacts with the two parallel orientated ones, which results in lower energy than interaction with only one chain. Therefore, the optimized bundle structures in the C50-range are ~100 kJ mol-1 lower in energy than the all-trans conformers. This result is in qualitative agreement with the results of Thomas and coworkers [5].
A fundamental advantage of molecular quantum chemistry tools such as MedeA MOPAC is their ability to compute the standard entropy and ideal heat capacity from the vibrational analysis of any stable conformer, while at the same time keeping the computational effort low. This capability opens the way to an understanding of the distribution at various temperatures and thermodynamic properties of conformers.
With MedeA, we now build triglycerides in an extended configuration (separate all-trans chains). From the structural optimization, we note that the most stable forms are folded conformers with three parallel chains interacting. Fewer gauche dihedral angles are necessary than with alkanes, and they are related to the same branch. Up to 57 carbon atoms (i.e., 18 C atoms per chain), the standard enthalpy of formation of folded triglycerides determined with MOPAC-PM7 follows a decreasing trend with increasing length. Similarly to linear alkanes, folded configurations are favored overextended star conformers when the length of all-trans segments increases.
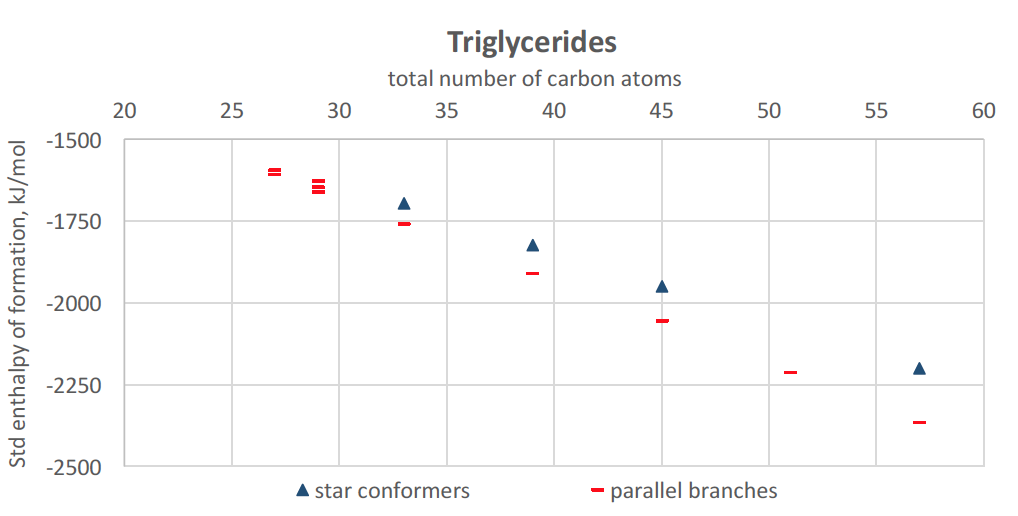
Figure 27.5.2 Standard enthalpy of formation of selected conformers of triglycerides (27 to 58 carbons), using MedeA MOPAC. Red bars: folded conformations with parallel branches, Blue triangles: star conformations (arms pointing at three different directions).
27.6. Conclusion and outlook
MedeA MOPAC correctly and consistently describes the folded stable conformers of long-chain alkanes and triglycerides. The trends for enthalpies of formation computed with MOPAC for chain lengths and folding are in line with literature results obtained with ab-initio quantum chemistry methods.
PM7, as available in MOPAC, is very useful in analyzing large numbers of stable conformers of large molecules and in providing a theoretical reference state in the thermodynamics of solvation or the modeling of liquid-vapor equilibria. We can use this approach to compute the free energy of any stable conformer, or in other words, to obtain standard free energies accounting for entropic effects [11]. Standard free energies, in turn, are needed for the determination of conformer concentrations, as was shown for alkanes [6].
Further promising application areas include the study of solvent effects and describing the influence of temperature on conformer distributions. In this context, the combination of high-productivity software tools with high-level theory (ab initio methods with extended basis sets), and validated semiempirical methods allow for a systematic description of larger molecules. In combining these elements, MedeA MOPAC is well-positioned to help to understand the behavior of many types of organic compounds and to guide experiments on phase transitions, reactions, and transport properties.
| [1] | A conformer is an isomer of a molecule differing from other conformers by rotation around one or more covalent single bonds. |
| [2] | James J. P. Stewart, “Stewart Computational Chemistry”, Colorado Springs, CO, USA, HTTP://OpenMOPAC.net (2016) |
| [3] | MedeA and Materials Design are registered trademarks of Materials Design, Inc. |
| [4] | (1, 2, 3) J. J. P. Stewart, “Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO approximations and Re-Optimization of Parameters”, J. Mol. Model. 19, 1 (2013) (DOI) |
| [5] | (1, 2, 3, 4, 5, 6) L. L. Thomas, T. J. Christakis, and W. L. Jorgensen, “Conformation of Alkanes in the Gas Phase and Pure Liquids”, J. Phys. Chem. B 110, 21198 (2006) (DOI) |
| [6] | (1, 2, 3, 4) N. O. B. Luettschwager, T. N. Wassermann, R. A. Mata, and M. A. Suhm, “The Last Globally Stable Extended Alkane”, Angew. Chem., Int. Ed. 52, 463 (2013) (DOI) |
| [7] | B. P. Tissot, and D. H. Welte, “Petroleum formation and occurrence”, 2nd edition, Springer Verlag, Berlin, (1984) |
| [8] | (1, 2) L. H. Wesdorp, “Liquid-Multiple Solid Phase Equilibria in Fats: Theory and Experiments”, PhD memoir, TU Delft (1990) (DOI) |
| [9] | D. A. McQuarrie, (1976), “Statistical Mechanics”, New York, Harper and Collins, New revised edition, (2000) |
| [10] | D. Weininger, “SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules”, J. Chem. Inf. Comput. Sci. 28, 31–36 (1988) (DOI) |
| [11] | X. Rozanska, J. J. P. Stewart, P. Ungerer, B. Leblanc, C. Freeman, et al., “High-Throughput Calculations of Molecular Properties in the MedeA Environment: Accuracy of PM7 in Predicting Vibrational Frequencies, Ideal Gas Entropies, Heat Capacities, and Gibbs Free Energies of Organic Molecules”, J. Chem. Eng. Data 59, 3136 (2014) (DOI) |
Required Modules
- MedeA Environment including the Molecular Builder, Conformer Search, analysis tools, and job control (JobServer and TaskServer)
- MedeA MOPAC and MedeA MOPAC GUI
- MedeA Gaussian and MedeA Gaussian GUI
| download: | pdf |
|---|