3. Colloidal behavior of confined model asphaltenes using MD Simulations
3.1. Introduction
The colloidal behavior of asphaltenes explains the high viscosity of heavy oils and the occurrence of solid deposits in reservoir rocks, production wells or transport lines.
Asphaltenes make refining more difficult because they cause coke deposits and tend to lower the yield in high-quality fuels or chemicals. Due to their significant content of sulfur, the existence of traces of metals (i.e. Ni, V) and their high aromaticity, energy-intensive conversion and hydrotreatment processes are required.
Operationally, crude oil asphaltenes are defined as being soluble in aromatic solvents such as benzene, toluene, pyridine and marginally soluble in n-alkanes [1]. From a structural viewpoint, asphaltenes are poly-aromatic molecules bearing a variable number of different alkyl chains and saturated cycles. Elemental analysis shows that they are significantly poorer in hydrogen than crude oils (H/C = 0.9 to 1.3, [2]) and comprise significant amounts of sulfur, oxygen, and nitrogen. As shown recently by Atomic Force Microscopy (AFM) and scanning tunneling microscopy (STM) of asphaltenes [3] their polyaromatic cores present a large range of possible structures.
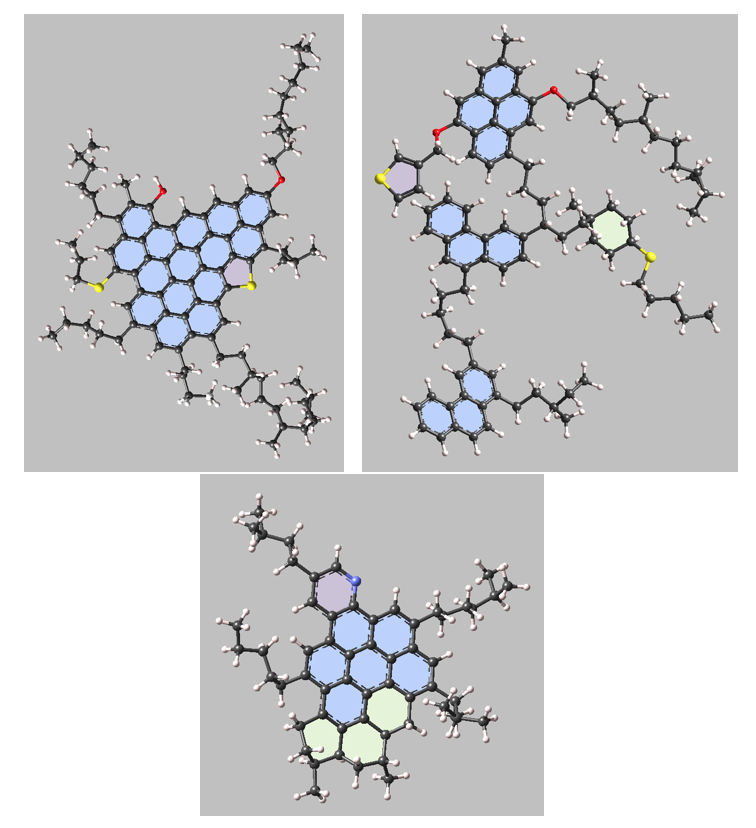
Figure 3.1.1 Examples of molecular models of asphaltenes. (Top-left) continental model, (Top-right) archipelago model, (Bottom) island model.
The colloidal nature of asphaltenes is known experimentally [4], [5], [6]. It is generally explained by Yen’s model, in which 4-5 molecules form stacks of poly-aromatic sheets [5], [6]. However, the behavior of such colloids is still a subject of intensive research, mainly because of the difficulty to consistently determine the behavior of heavy oils under high-temperature conditions, as they are present in steam injection processes and under refining conditions.
Generally, conducting relevant experiments under extreme conditions is costly and difficult, and in particular, when aiming at studying asphaltene behavior in the presence of high-pressure gases in deep reservoirs, or when attempting to describe asphaltene interaction with porous media, solvents, and additives. The progress of analytical techniques has greatly improved the knowledge of asphaltene structure and diversity, but this triggers a need for property/structure relationships that are still poorly known for these macromolecules.
Keywords: asphaltenes, LAMMPS, molecular dynamics
3.2. Molecular Modeling
Molecular modeling can be used for improving understanding of asphaltenes [7], [8], [9]. As illustrated by Materials Design using the MedeA modeling environment [10], atomistic simulations are very well suited to model industry-relevant scenarios involving the colloidal behavior of asphaltene-containing systems.
As an illustration, we compare the colloidal behavior of three asphaltene models of similar molecular weight (Figure 3.1.1). in two liquid solvents (toluene and n-heptane), using MedeA LAMMPS [11] to perform Molecular Dynamics simulations with the All-Atom pcff+ forcefield.
Similar to forcefields developed for the Nobel-winning investigation of biomolecules [12], pcff+ is a large coverage / high quality forcefield, suitable for the accurate description of fluid properties.
Asphaltene nano-aggregation or dissociation takes place on a time scale of typically a few nanoseconds. Molecular Dynamics simulations were conducted for up to 15 ns, a simulation time that is comparable to previous work [9].
Snapshots of the three different systems at equilibrium (using a different asphaltene model in each system and toluene as the solvent) are shown in Figure 3.2.1, Figure 3.2.2 and Figure 3.2.3.
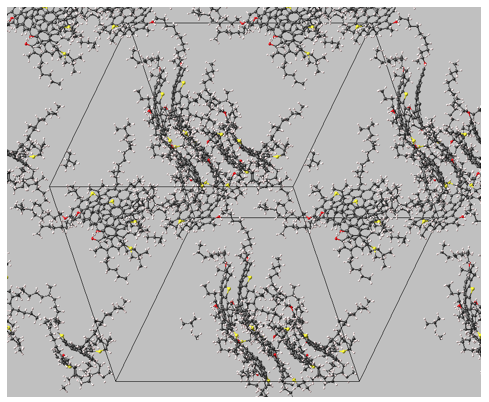
Figure 3.2.1 Conformations of 1.5 mol % of asphaltenes (continental model) in toluene at 350 K, 1 bar (solvent molecules not shown). Three clusters comprising four, two and two stacked polyaromatic units with ca. 0.37 nm spacing between aromatic planes.
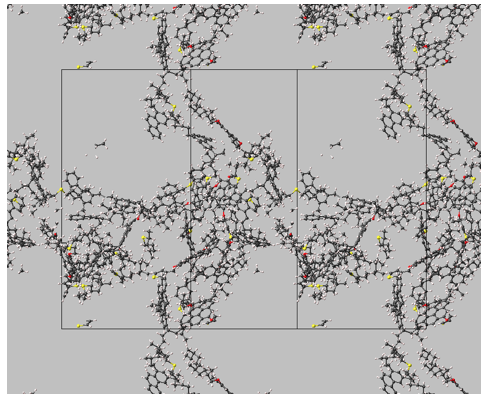
Figure 3.2.2 Conformations of 1.5 mol % of asphaltenes (archipelago model) in toluene at 350 K, 1 bar (solvent molecules not shown). Parallel stacking is limited.
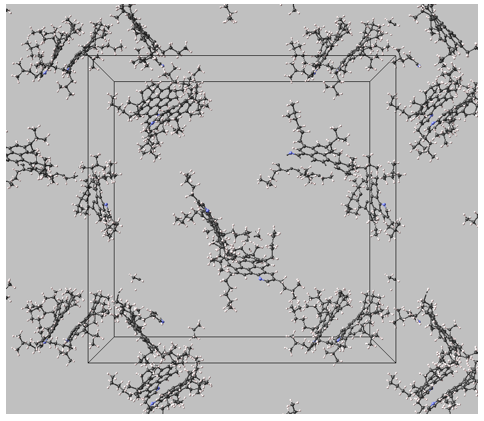
Figure 3.2.3 Conformations of 1.5 mol % of asphaltenes (island model) in toluene at 350 K, 1 bar (solvent molecules not shown). Island model, stacking is observed but limited to a maximum of two polyaromatic units.
3.3. Results
The degree of aggregation can be monitored by analysis of the molecular dynamics trajectories as shown in (Figure 3.3.1).
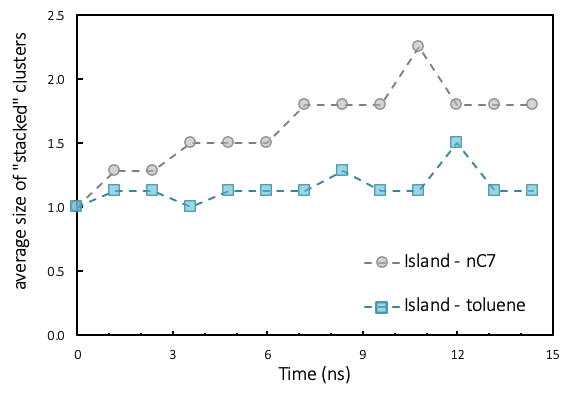
Figure 3.3.1 “Stacked” clusters (aggregates formed by stacked polyaromatic units) of island asphaltenes in toluene and in n-heptane.
The aggregation analysis shows that the island model agrees with the definition of asphaltenes, as it provides an explanation of the stronger nano-aggregation in n-heptane than in toluene and the limited size of nano-aggregates (Figure 3.3.1). The growth of nano-aggregates is limited beyond four molecules, because of steric repulsion of lateral alkyl chains and bending of poly-aromatic planes.
With MedeA LAMMPS, the effect of gravity acting on the system was modeled, thus providing a way to investigate the conditions in which a heavy liquid phase can segregate (Figure 3.3.2).
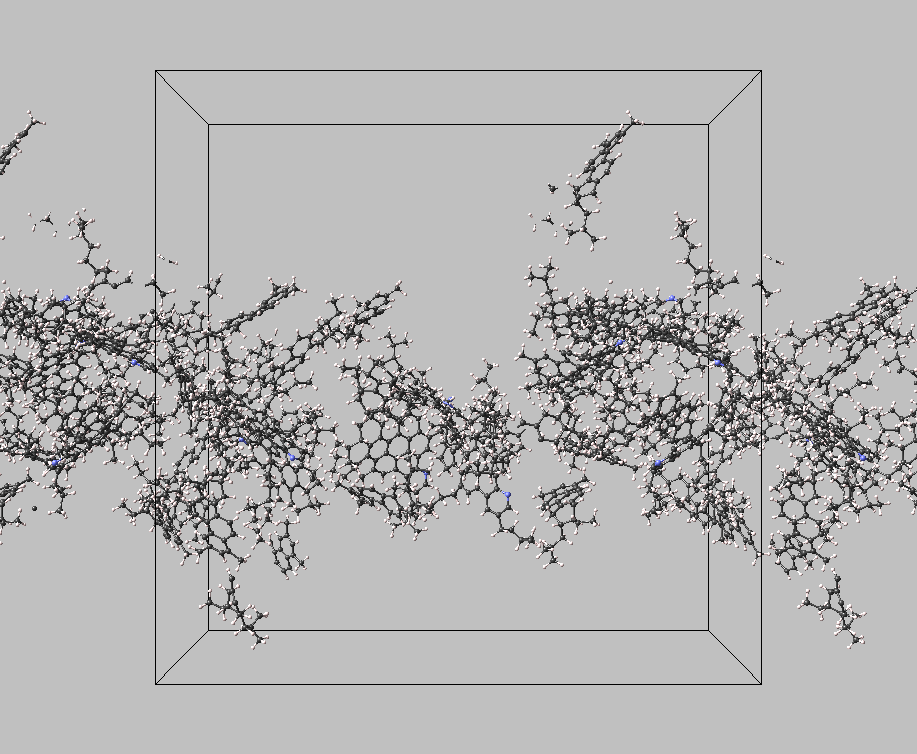
Figure 3.3.2 Conformation of a mixture of asphaltenes, model resins in liquid n-hexane at 473 K. A gravity-like force has been applied for a period of 1 ns with MedeA LAMMPS, to illustrate the gathering of a bitumen-like phase in the middle of the simulation box. Model resins are smaller than asphaltenes, as they comprise four aromatic rings and fewer alkyl chains in this example.
The use of the radial distribution functions to further investigate stacking distances, as shown in (Figure 3.3.3), is also a powerful option.
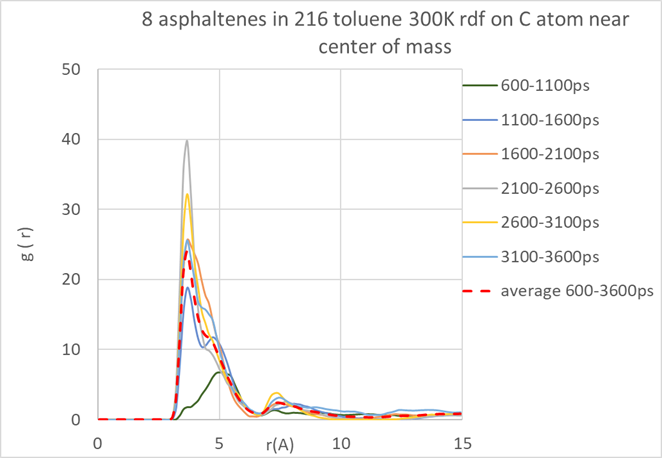
Figure 3.3.3 Radial distribution function for tagged carbon atoms near the center of mass of the asphaltene molecules, calculated over different simulation time periods.
The current implementation in MedeA opens the door for further in-depth studies:
- understanding the role of resins (Figure 3.3.1), using additional molecular simulation techniques (MedeA VASP for interactions with mineral surfaces, MedeA GIBBS for adsorption and gas solubility [13], coarse-grained models for large scale or long times),
- determining properties of asphaltenes dispersed in solvents, such as the asphaltenes’ apparent density.
Oil industry (upstream and downstream) can greatly benefit from the use these tools, along with traditional experimental techniques, in order to better understand and control the macroscopic behavior of asphaltenes and its molecular origin.
| [1] | G. J. Speight, “Petroleum Asphaltenes - Part 1: Asphaltenes, Resins and the Structure of Petroleum”, Oil & Gas Science and Technology 59, p. 467-477 (2004) doi.org/10.2516/ogst:2004032 |
| [2] | E. Rogel et al., “Sediment Formation in Residue Hydroconversion Processes and Its Correlation to Asphaltene Behavior” Energy & Fuels 27 p. 6587 (2013) doi.org/10.1021/ef401614a |
| [3] | B. Schuler et al., “Unraveling the Molecular Structures of Asphaltenes by Atomic Force Microscopy”, J. Am. Chem. Soc. 137, p. 9870 (2015) doi.org/10.1021/jacs.5b04056 |
| [4] | D. Fenistein et al., “Viscosimetric and Neutron Scattering Study of Asphaltene Aggregates in Mixed Toluene/Heptane Solvents”, Langmuir 14, p. 1013 (1998) doi.org/10.1021/la9709148 |
| [5] | (1, 2) I. Merdrignac et al., “Physicochemical Characterization of Petroleum Fractions: the State of the Art”, Oil & Gas Science and Technology 62, p. 7 (2007) doi.org/10.2516/ogst:2007002 |
| [6] | (1, 2) O. C. Mullins et al., “Advances in Asphaltene Science and the Yen–Mullins Model”, Energy & Fuels 26, p. 3986 (2012) doi.org/10.1021/ef300185p |
| [7] | L. Zhang et al., “Molecular Orientation in Model Asphalts Using Molecular Simulation”, Energy & Fuels 21, p. 1102 (2007) doi.org/10.1021/ef060449z |
| [8] | L. Zhang et al., “Relaxation time, diffusion, and viscosity analysis of model asphalt systems using molecular simulation”, J. Chem. Phys. 127, p. 194502 (2007) doi.org/10.1063/1.2799189 |
| [9] | (1, 2) T. F. Headen et al., “Evidence for Asphaltene Nanoaggregation in Toluene and Heptane from Molecular Dynamics Simulations”, Energy & Fuels 23, p. 1220 (2009) doi.org/10.1021/ef800872g |
| [10] | P. Ungerer et al., “Sensitivity of the aggregation behaviour of asphaltenes to molecular weight and structure using molecular dynamics”, Mol. Sim. 40, p. 115 (2013) doi.org/10.1080/08927022.2013.850499 |
| [11] | S. Plimpton, “Fast Parallel Algorithms for Short-Range Molecular Dynamics”, J. Comput. Phys. 117, p. 1 (1995) doi.org/10.1006/jcph.1995.1039 |
| [12] | A. D. MacKerell et al., “All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins”, J. Phys. Chem. B 102, p. 3586 (1998) doi.org/10.1021/jp973084f |
| [13] | M. Yiannourakou, P. Ungerer, B. Leblanc, X. Rozanska, P. Saxe, S. Vidal-Gilbert, F. Gouth and F. Montel, “Molecular Simulation of Adsorption in Microporous Materials”, Oil & Gas Science and Technology 68, p. 977 (2013) https://doi.org/10.2516/ogst/2013134 |
Required Modules
- MedeA Environment including the modules Molecular Builder, LAMMPS, LAMMPS GUI, Flowcharts, Forcefield Bundle and job control (JobServer and TaskServer)
- MedeA Amorphous Materials Builder
| download: | pdf |
|---|