9. Diffusion of Hydrogen in Nickel
Mass transport is of central importance in the processing, operation, and aging of many materials. However, despite the importance of diffusion, the experimental measurement of diffusion coefficients is complicated by the superposition of competing transport mechanisms. Defects and impurities (for example vacancies and oxygen impurities in metals), surface migration, and intergranular transport modes may all be present in a given system. The deconvolution of the underlying origins of observed transport phenomena can provide substantial understanding and insights. Computational techniques have now matured to the point where calculated temperature-dependent diffusion coefficients yield an accuracy and reliability comparable with experiments for diffusion properties. The present application illustrates the direct computation of hydrogen diffusion rates as a function of temperature in metallic nickel.
Keywords: diffusion in solids, hydrogen, nickel, computations, DFT, VASP
9.1. Rationale
The present study illustrates the accuracy and reliability of computed properties such as the diffusion coefficients of atoms in solids. A substantial number of diffusion coefficients for hydrogen in a range of pure metals have been reported in the literature. However, for many systems the discrepancies between differing experimental data can span several orders of magnitude. The case of hydrogen in nickel, H in Ni, is however well characterized and has been selected here as a benchmark system, providing a direct test of the ability of density functional theory to describe not only ground state but also transition state energies and geometries.
9.2. Methodology
To this end, accurate DFT calculations were carried out with MedeA®[1] VASP using the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional. Importantly, the present approach includes temperature-dependent vibrational contributions (using MedeA Phonon) to the Gibbs free energy as well as effects due to thermal expansion. With this rigorous computational approach it is possible to assess the accuracy of computated diffusion coefficients for systems such as H in Ni.
9.3. Results
The computed temperature-dependent diffusion coefficient of H in Ni compares extremely well with experimental data as illustrated in Figure 9.3.1. The agreement is particularly impressive near room temperature, where experimental data are scattered and where the predicted diffusion coefficient is at the center of the experimental observations. At elevated temperatures the measured diffusivity is slightly lower than the computed values. It is worth noting that the computation employs a straightforward harmonic model and does not explicitly include the effects of anharmonicity and trapping of diffusing species, effects that are likely to be of increasing importance at high temperature.
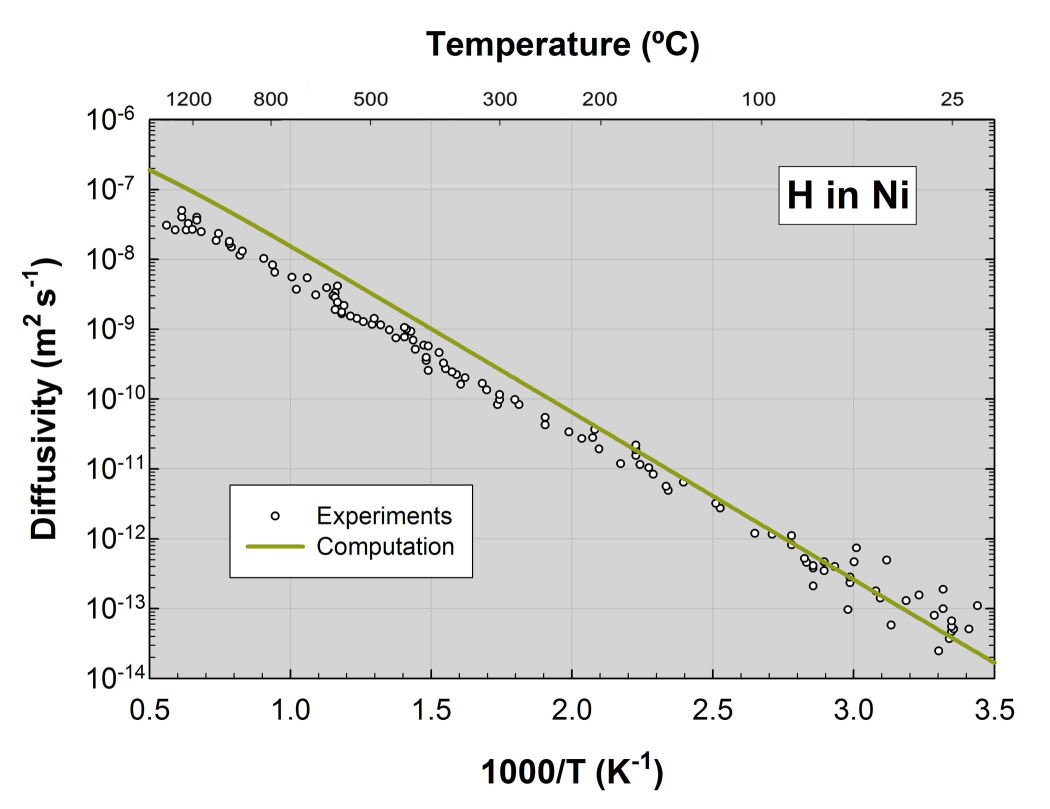
Figure 9.3.1 Computed and experimental diffusion coefficient of H in Ni. See Ref. [2] for detailed explanations.
9.4. Significance
The mass diffusion coefficient of atoms in solids can be computed with first-principles methods reaching an accuracy which can be comparable with experiment. Computational methods provide reliable values for diffusion coefficients for systems and conditions where experiments are difficult, time consuming, or costly, for example in the aging of materials. Furthermore, insight into the mechanisms of the rate limiting steps in a diffusion process provides valuable guidelines for the improvement of novel materials such as fast solid state ion conductors for batteries and fuel cells.
MedeA modules used in this application
- MedeA Environment
- MedeA VASP
- MedeA Phonon
- MedeA Transition State Search
9.5. Comments
A critical step in the calculation of diffusion coefficients is the choice of the relevant mechanism and diffusion pathways. In the present case, symmetry dictates the necessary diffusion path. In more complex systems, the determination of the diffusion mechanism requires analysis of competing pathways. The nudged elastic band method (as provided by MedeA Transition State Search) allows for the exploration of the diffusion pathways present in a given system. For relatively fast processes, e.g. diffusion at elevated temperature, diffusion coefficients can be obtained directly from molecular dynamics simulations using MedeA LAMMPS and the MedeA Diffusion module, with appropriate forcefields. Additionally, the MedeA Forcefield Optimizer allows the construction and optimization of forcefield parameters based on MedeA VASP derived first-principles results.
| [1] | MedeA and Materials Design are registered trademarks of Materials Design, Inc. |
| [2] | E. Wimmer, W. Wolf, J. Sticht, P. Saxe, C. B. Geller, R. Najafabadi, and G. A. Young, “Temperature-dependent diffusion coefficients from ab initio computations: Hydrogen, deuterium, and tritium in nickel”, Phys. Rev. B 77, 134305 (2008) |
| download: | pdf |
|---|